

Abstract
Grand canonical Monte Carlo (GCMC) simulation combined with the ideal adsorbed solution theory (IAST) and a statistical method were utilized to investigate the effect of functional groups on zirconium oxide based metal-organic frameworks (MOFs) Zr6-AzoBDC (Zr6A) for the gases (H2, CH4) adsorption property and CO2/CH4 selectivity under low pressure. The results showed that phenyl groups containing nitrogen (pyridine, pyrimidine) and thiophene group enhance the gas affinity with MOFs, therefore increasing both gravimetric and volumetric uptake. In addition, this behavior can also cause significantly improved selective capture of CO2 from CO2/CH4 gas mixtures. Among functional groups studied, the sulfonic acid group can potentially improve CH4, CO2 uptake and H2 isosteric heat of adsorption. These findings would play a vital role in designing new materials toward gas adsorption properties.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
It is no exaggeration to state that one of the biggest challenges for human beings in the 21st century is global warming and energy crisis. The anthropogenic emission of CO2 from industrial processes has greatly affected the environment, and this origin of pollution, together with other troubling issues, is a major concern. In addition, the present oil reserves have been drastically reduced, so there is no doubt that the supply of fossil fuel could be exhausted in the near future. It has become necessary to seek out new sources of energy, such as solar energy, wind power, hydropower and so forth. Amongst those candidates, energy from H2 or CH4 has received a lot of attention due to their negative greenhouse effects. An important challenge of using these gases as an energy sources is an efficient storage system. Numerous studies have been conducted to improve the current technology. During the past decades, metal-organic frameworks (MOFs) have shown a huge potential for the challenges mentioned above. Indeed, MOFs are porous, chemically diverse, and structurally designable materials; most importantly, they are extremely versatile. These porous materials are promising for gas storage and separation as well as many other potential applications. However, the major issue of these promising materials is often sensitivity to moisture, and the structure can completely collapse with the introduction of water vapor. Fortunately, Zr-based MOFs have been proved as thermo-chemically and structurally stable compounds, irrespective of the presence of water molecules [1]. The ability of Zr-MOFs to retain the porous structure under humid conditions has attracted our attention in the quest for a practical material for storage and separation application.
We herein report a theoretical investigation on the effect of functional groups on Zr6-AzoBDC (Zr6A) in order to enhance H2, CH4 storage and CO2 capture. The grand canonical Monte Carlo (GCMC) simulation combined with the ideal adsorbed solution theory (IAST) [2] is employed. Our proposed compounds were designed by means of a post-synthetic modification (PSM) method [3], i.e. the altering of the chemical composition of the linker without changing its underlying net, which is known as an efficient approach in experimentally synthesizing new MOFs. Our calculations demonstrate that the PSM approach coupled with theoretical calculations is a strategic method to improve the adsorption properties of these materials.
2. Computational details
2.1. Grand canonical Monte Carlo simulations
All grand canonical Monte Carlo (GCMC) simulations were carried out using MUSIC software package [4]. The interaction between the adsorbent (MOF) with methane (CH4) and carbon dioxide (CO2) were described mainly by van der Waals forces. The electrostatic forces in this case do not play an important role. We used the Lennard–Jones model with the parameters obtained from the transferable force fields (TraPPE) for molecular gas (adsorbate) and Dreiding force field [5] for the atoms in the MOF (adsorbent). The Lorentz–Berthelot rule was used to calculate the parameters for interaction between gas molecules and the MOF. The Lennard–Jones interactions of distances greater than 12.8 Å are ignored. In the simulation, a supercell 2 × 2 × 2 (i.e. 8 unit cells) of MOF was kept rigid, the molecular gas was considered a 'spherical molecule'. Each point of the isotherm was obtained by 15–20 million simulation steps. The authenticity of this methodology has been proven by reported literature [6]. In the view of adsorption theory, one needs to distinguish the nature of simulation with that of the experiment. While the result calculated by GCMC simulation is the amount of gas molecules in the pore of adsorbent, namely the total amount Nabs, current experimental techniques of are not able to characterize this absolute amount of adsorbed molecules. Fundamentally, these measurements just produce the difference in the amount of total adsorbed gas and the amount of bulk gas at same condition of measurement, namely Nex. The equation to converse between two these quantities was given by [7]
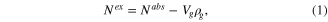
where Nex, Nabs excess and absolute amount, respectively, Vp is pore volume of the adsorbent and ρg is the fluid density in the bulk phase at the same temperature and pressure for adsorption, which is calculated by the Peng–Robinson equation of state [8]. At low pressure (lower than 1 atm) the difference between Nex and Nabs is negligible.
2.2. Molecular mechanics
Molecular mechanics (MM) is a practical technique to study atomistic systems containing thousands of atoms per unit cell. Total potential energy of the system is determined for each set of positions of the atoms by using intermolecular/intramolecular potential function in the classical force field. It helps to refine the repetitive geometry of a mechanical approach until some predetermined criterion of convergence is satisfied. Finally, the quality of geometrical optimization depends on the accuracy of the force field. Amongst the diversity of force fields developed, the UFF force field is probably the most versatile since its parameters are derived for most of the atoms in the periodic table. In addition, this force field can combine with the QEq method to study systems in which electrostatic interactions [9] are important. Moreover MM method possesses an advantage in optimizing systems, such as MOF-205, PCN-14, MIL-101(Cr) [10] since the implement of high computational-cost calculations are not reasonably practicable. In this study, MOF structures are optimized with Dreiding force fields and UFF using GULP software [11].
2.3. Adsorption enthalpy calculation
The isosteric heat of adsorption [12] (Qst) is based on the thermochemical parameters of the adsorption process. Adsorption enthalpy (ΔHads) can be calculated from gas adsorption isotherms simulated at two or more different temperatures by means of fitting of the virial equation. The zero-coverage isosteric heat corresponds to the interaction energy between gas molecule and the strongest interaction site of the MOF. The virial equation
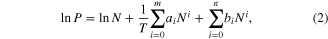
where P is pressure, N is the amount adsorbed CH4 gas, T is temperature, m and n represent the numbers of coefficients required to adequately describe the isotherms. The virial equation consists of temperature-independent parameters ai and bi is used to fit the sorption data. Adsorption isotherms measured at 273 K and 298 K are used in this procedure by applying the statistical program Origin 8.5 (MicroCal Software Inc., Northampton, MA). ΔHads is then calculated by following equation
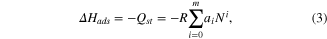
where R is universal gas constant.
2.4. Adsorption selectivity
Adsorption-based separation is a physisorptive operation governed by thermodynamic equilibrium processes, which relies on the fact that guest molecules reversibly adsorb in nanopores at densities that far exceed the bulk density of the gas sources in equilibrium with the adsorbents [13].
It has been shown in numerous published papers [14–17] that the IAST can be used to estimate quite accurately adsorption equilibrium of mixtures from pure component isotherm data. For a binary gas mixture adsorption (A, B components), the predicted adsorption selectivity (SA/B) was calculated by
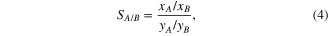
where xA, xB and yA, yB are the mole fractions of A and B in the adsorbed and bulk phases, respectively.
3. Results and discussion
Based on Zr6-AzoBDC (Zr6A) material synthesized by Yang et al [18, 19], the studied structures was optimized with the Dreiding force field and UFF as implemented in the GULP code. GCMC simulation was then performed to calculate the H2, CH4 and CO2 adsorption isotherms. To interpret the effects of the functional group on Zr6A, nine new MOF models with different substituents were introduced, namely, Zr6ACl, Zr6AC6, Zr6AC5N, Zr6AC4N2, Zr6AC4N, Zr6AC4O, Zr6AC4S, Zr6AC5 and Zr6ASO3H [20]. These materials are the products of incorporating a certain substituent into the Azo-linker (figure 1) and their CO2/CH4 selectivity was investigated by means of IAST.
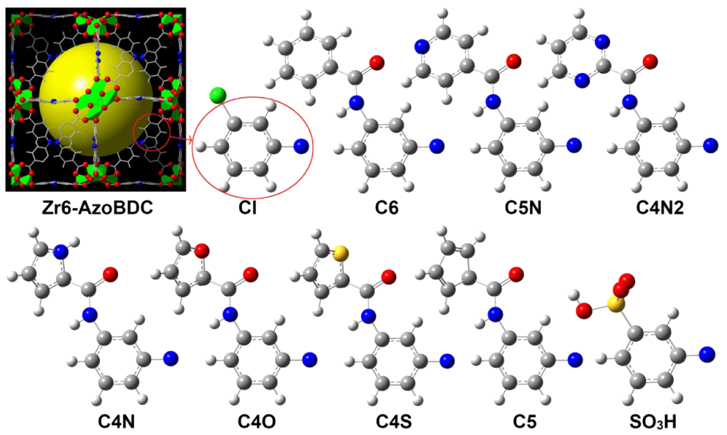
Figure 1. Model of Zr6-AzoBDC structure and functional groups: The large spheres represent the void regions inside the cages (Zn polyhedral: blue for octahedral cage; C, gray; O, red; N, blue; Cl, green; S, yellow). The extended organic linkers attached on Zr6-AzoBDC are also presented.
Download figure:
Standard image High-resolution imageThe volumetric uptake and isosteric heats of adsorption of MOFs are presented in figures 2, 3 and 4, in which, H2 isotherm is calculated in units of mg/g, while both CH4, CO2 isotherms are calculated in c.c./c.c. units.

Figure 2. H2 isotherm at 77 K (a), 87 K (b) and isosteric heat of adsorption (c) at 1 atm.
Download figure:
Standard image High-resolution image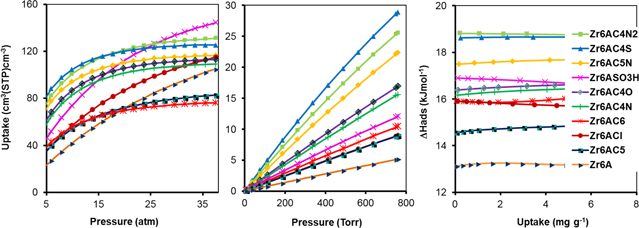
Figure 3. CH4 isotherm at high pressure (a), low pressure (b) and isosteric heat of adsorption (c) at 298 K.
Download figure:
Standard image High-resolution image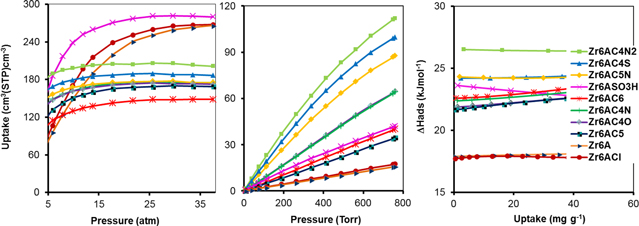
Figure 4. CO2 isotherm at high pressure (a), low pressure (b) and isosteric heat of adsorption (c) at 298 K.
Download figure:
Standard image High-resolution image3.1. Adsorption isotherms of hydrogen (H2), methane (CH4) and carbon dioxide (CO2)
Hydrogen (H2) uptake of Zr6AC5N, Zr6AC4N2 and Zr6AC4S are 24.06, 23.69 and 23.73 mg g−1, respectively; these values are higher than those of unmodified Zr6-AzoBDC (13.55 mg g−1) and other MOFs at 77 K and 1 atm. This may be because Zr6AC5N, Zr6AC4N2 and Zr6AC4S contain nitrogen phenyl group such as pyridine, pyrimidine and thiophene. The nitrogen and sulfur atoms possess a high affinity with gas, thereby improving their isosteric heat of adsorption. Furthermore, this may be due to the sulfonic acid group, corresponding to Zr6ASO3H, which significantly enhanced the isosteric heat of adsorption (8.39 kJ mol−1) at 87 K and 1 atm (table 1) and this result is consistent with previous research [21, 22].
Table 1. Summary of porosity, H2 (at 1 atm, 77 K), CH4 and CO2 uptake (at 38 atm, 298 K), and enthalpy of adsorption for materials in this study.
| Material | ASABET (m2 g−1) | Vporea (cm3 g−1) | VH2b (mg g−1) | ΔHads,H2d (kJ mol−1) | VCH4c (c.c. c.c.−1) | ΔHads,CH4d (kJ mol−1) | VCO2c (c.c. c.c.−1) | ΔHads,CO2d (kJ mol−1) |
|---|---|---|---|---|---|---|---|---|
| Zr6A | 4744 | 1.31 | 13.55 | 4.72 | 105.09 | 13.13 | 266.28 | 17.91 |
| Zr6ACl | 3569 | 1.09 | 13.94 | 6.76 | 115.28 | 15.92 | 267.57 | 17.70 |
| Zr6AC6 | 1025 | 0.38 | 19.67 | 6.89 | 76.14 | 15.91 | 148.30 | 22.59 |
| Zr6AC5N | 1182 | 0.46 | 24.08 | 6.76 | 116.48 | 17.48 | 175.08 | 24.34 |
| Zr6AC4N2 | 1426 | 0.56 | 23.69 | 7.91 | 131.31 | 18.82 | 201.35 | 26.54 |
| Zr6AC4N | 1334 | 0.47 | 19.83 | 6.54 | 109.12 | 16.16 | 175.14 | 22.37 |
| Zr6AC4O | 1337 | 0.49 | 20.25 | 6.50 | 113.03 | 16.38 | 172.35 | 21.86 |
| Zr6AC4S | 1258 | 0.49 | 23.73 | 6.61 | 125.43 | 18.62 | 186.63 | 24.22 |
| Zr6AC5 | 1423 | 0.44 | 18.05 | 6.08 | 82.70 | 14.55 | 168.83 | 21.65 |
| Zr6ASO3H | 2699 | 0.89 | 17.66 | 8.39 | 144.50 | 16.90 | 297.77 | 23.65 |
apore volume, bgravimetric uptake, cvolumetric uptake, dadsorption heat at zero coverage calculated from the virial equation.
Similarly, the Zr6ASO3H, Zr6AC4N2 and Zr6AC4S (144.50, 131.31 and 125.43 c.c. c.c.−1) show higher methane volumetric uptakes than unmodified Zr6-AzoBDC (105.09 c.c. c.c.−1) and other MOFs at 298 K and 38 atm (table 1). This is explained by the high affinity with the gas of nitrogen and sulfur atoms. Also the isosteric heats in methane adsorption of these materials are improved (Zr6AC4N2: 18.82 kJ mol−1 and Zr6AC4S: 18.62 kJ mol−1, compared with 13.13 kJ mol−1 of Zr6-AzoBDC). In contrast, Zr6AC6 and Zr6AC5 possess the lower capacity of methane adsorption, corresponding to the volumetric uptakes of 76.14 and 82.70 c.c. c.c.−1, respectively.
For CO2 adsorption, Zr6ASO3H shows the highest volumetric uptake (297.77 c.c. c.c.−1 at 298 K, 38 atm) and Zr6AC4N2 has the highest isosteric heat of adsorption (26.54 kJ mol−1 at 298 K and 1 atm) (table 1). They have significantly enhanced volumetric uptake and absorption heat energy compared to unmodified Zr6-AzoBDC (266.28 c.c. c.c.−1 and 17.91 kJ mol−1) as well as compared to other groups, since the sulphonic acid group increases both the heat of adsorption and total uptake for carbon dioxide.
3.2. Adsorption selectivity (CO2/CH4)
According to gas selectivity studies, the initial slopes for CO2 and CH4 adsorption uptakes indicate noticeable affinities for CO2/CH4 of proposed structures at high pressure. We studied the selectivity adsorption by the ideal adsorbed solution theory (IAST), which calculates the system's gas selectivity capabilities of theoretical gas mixtures utilizing the pure component isotherms in figures 3 and 4, and the results are shown in figure 5. It should be noted that even though IAST calculations are performed using GCMC isotherm, their selectivity results represent theoretical values that might deviate from practical applications. The selectivity for CO2/CH4 of new MOFs is quite significant at 22 bar for 50/50 mixture of CO2 and CH4. The selectivity of MOFs that contain C4N2 and C5N (pyridine and pyrimidine) groups would increase with increasing pressure; while the Zr6ACl and Zr6AC6 decreased at high pressure. The validity of IAST calculations is dependent on the ideality of MOFs [23]. We confirm this result by calculating selectivity from the initial slopes of the isotherms (figure 6). The resulting selectivity for CO2/CH4 are in agreement with the IAST value. In summary, the pyridine and pyrimidine groups can enhance CO2/CH4 selectivity.
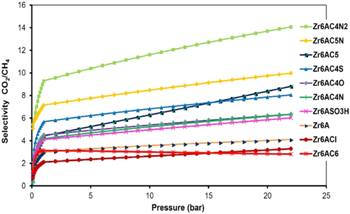
Figure 5. Simulation results for separation of an equimolar mixture of CO2/CH4 in new MOFs at 298 K.
Download figure:
Standard image High-resolution image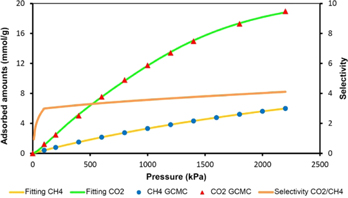
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. The IAST-predicted isotherms and selectivity's of equimolar mixture of CO2 and CH4 in Zr6-AzoBDC at 298 K.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusion
The functionalization of Zr6-AzoBDC can remarkably improve H2, CH4 uptake and CO2/CH4 separation. The substituents containing phenyl group with nitrogen inside and sulfur atoms such as pyrazine, pyridine and thiophene groups show a better performance than other groups for gas storage and separation. This can be explained by the increase of isosteric heat of adsorption. In particular, the sulfonic acid group has significantly enhanced both the gas storage capacity and isoteric heat of adsorption. The GCMC simulation demonstrates itself a beneficial tool to aid experimental chemists in designing new promising nano and micro porous materials.
Acknowledgments
The authors gratefully acknowledge SR16000 supercomputing resources from the Center for Computational Materials Science of the Institute for Materials Research, Tohoku University, Japan and the Institute for Computational Science and Technology (ICST), Ho Chi Minh City for their support.