


Abstract
To evaluate the cytotoxic effect of chitosan nanoparticles (CS-NPs) on an in vitro human liver cancer cell model (HepG2) and their possible application as a drug delivery system, we synthesized water-soluble CS-NPs, investigated their properties and extensively evaluated their cytotoxic activity on the cellular and molecular levels. A human liver cancer cell line was used as a model of human liver cancer. The CS-NPs were characterized using transmission electron microscopy, Fourier transform infrared spectroscopy, and zeta analysis. The cytotoxic effects of the CS-NPs on HepG2 cells were monitored by sulforhodamine B colorimetric assays for cytotoxicity screening and flow cytometric analysis. Molecular investigations including DNA fragmentation and the expression of some apoptotic genes on the transcriptional RNA level were conducted. Treatment of HepG2 with different concentrations of 150 nm diameter CS-NPs did not show alteration of cell morphology after 24 h of cell exposure. Also, when cells were treated with 100 μg ml−1 of CS-NPs, 12% of them were killed and IC50 reached 239 μg ml−1 after 48 h of cell exposure. Flow cytometry evaluation of the CS-NPs revealed mild accumulation in the G2/M phase followed by cellular DNA fragmentation after 48 h of cell exposure. Extensive evaluation of the cytotoxic effect of the CS-NPs showed messenger RNA (mRNA) apoptotic gene expression (p53, Bak, Caspase3) after 24 h of cell exposure with no expression of the mRNA of the caspase 3 gene after 48 h of cell exposure, suggesting the involvement of an intrinsic apoptotic caspase-independent pathway by increasing the exposure time of 100 μg ml−1 of the CS-NPs. The engineered CS-NPs were controlled to a 150 nm size and charges of 40 mV and a concentration of 100 μg ml−1 revealed a genotoxic effect on HepG2 after 48 h of cell exposure through intrinsic apoptotic caspase-independent mechanisms. Further quantitative analysis on the molecular and protein levels is still required to confirm the impact of chitosan size and time on genotoxic effect before reaching a final conclusion and starting its biomedical application.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Hepatocellular carcinoma (HCC) is the most common type of cancer among the Egyptian population. It is increasing in parallel to hepatotropic viral infections and is expected to at least double in the next 20 years. There are many strategies for treating HCC but all of them are very expensive and time consuming and have several side effects. Therefore another approach is needed for managing such an aggressive type of cancer [1].
Chitosan (CS), a cationic polysaccharide generated commercially by partial deacetylation of chitin, is a biodegradable polymer useful in a variety of applications including biomedicine, pharmaceuticals, metal chelation, and food additives. In addition, the absence of harsh chemicals in the CS manufacturing process makes it attractive as a promising drug delivery system to increase tumor activity [2]. CS nanoparticle (CS-NP)-delivered drugs accumulate selectively in tumor, rather than normal, tissues because of an enhanced permeation and retention effect [3]. In addition to its physicochemical properties, surface modifications of CS play a crucial role in the cytotoxic profile and targeting of cancers that are characterized by rapid division and aggressive growth [4]. Jeon and Kim found that CS oligomers possess antitumor activity tested both in vitro and in vivo [5]. In vitro CS-NPs exerted vigorous cytotoxicity against a colon cancer cell line (Calo320), gastric cancer cell line (BGC823), and liver cancer cell line (BEL7402) [6] and HepG2 [7]. In vivo, CS-NPs also showed significant dose- and size-dependent antitumor activity against sarcoma-180 and hepatoma H22 in mice. These findings suggest their application as a novel class of drugs against HCC [2].
The present study was conducted to develop soluble CS-NPs and evaluate their cytotoxicity on a HepG2 cell line as an in vitro model of human liver cancer cells hoping to prepare a nano-vehicle to deliver anti-HCC drugs and anti-hepatitis C virus genotype 4 (HCV-4) drugs.
2. Materials and methods
Low molecular weight CS, tripolyphosphate pentasodium and glacial acetic acid were purchased from Sigma-Aldrich.
2.1. Preparation of CS-NPs
Preparation of CS-NPs was carried out based on the ionotropic gelation method of CS with tripolyphosphate (TPP) anions as described by Calvo et al [8] with minor modification of the positive charge of the amino groups of CS cross-linked with the negatively charged TPP anions. The parameters were fixed at pH 4 and a CS:TPP ratio of 4:1.
2.2. Characterization of CS-NPs
The particle size, shape and size distribution profile were determined using high-resolution transmission electron microscopy (TEM). A drop from a very dilute sample solution was deposited on an amorphous carbon-coated copper grid and left to evaporate at room temperature. Imaging was accomplished using a Joel JEM-2100 microscope (accelerating voltage 200 kV; Gatan Erlangshen ES500 digital camera) at the National Research Center.
A Nano ZS apparatus (Malvern Instruments, UK) was used to determine the zeta potential of chitosan nanoparticles [9]. The zeta potential was determined by electrophoretic light scattering studies performed in an aqueous solution and Smoluchowski software approximation was used to calculate the zeta potentials. The measurements were recorded at 25 °C and started after 2 min to allow the temperature to equilibrate.
2.3. Fourier transform infrared spectroscopy (FTIR)
Molecules with covalent bonds may absorb IR radiation and the absorption is quantized where only certain IR frequencies are absorbed. The energy associated with IR radiation is sufficient to cause the molecules to rotate and vibrate when possible and the absorbing molecule is elevated to a higher energy state. The energy needed to cause an electron transition to the rotational level is smaller than the energy needed to cause a transition to the vibrational level. When the IR energy is between 1.239 eV and 0.0124 eV (energy high), the molecule will be excited to a higher vibrational state and when the energy is less than 0.0124 eV (energy weaker), the molecule is excited to higher rotational states in the gas phase. Since each vibration level has multiple rotational levels associated with it, the IR spectrum of a liquid or solid sample consists of broad vibrational absorption bands and not narrow lines. The model of the FTIR spectrometer utilized in this work is Nicolet iS10 [10–12].
2.4. Cell culture
A HepG2 human liver cancer cell line was obtained from VACSERA (the Holding Company for Biological Products & Vaccines). The cells were cultured and maintained in RPMI 1640 media (Biowest) supplemented with 10% fetal bovine serum and antibiotics (2% penicillin–streptomycin 100 IU ml−1), and 0.5% Fungizone. The cells were maintained in a monolayer culture at 37 °C under a humidified atmosphere of 5% CO2. The cells were subcultured by trypsinization (0.025% trypsin and 0.0025% EDTA), and maintained in a tissue culture laboratory at the National Cancer Institute, Cairo University, Egypt, with cryogenic banking of low-passage cells to maintain the uniformity of cell properties throughout the study [13]. Cell number and viability were monitored by standard trypan blue dye exclusion procedures. The growth curves for HepG2 were determined under baseline conditions prior to investigation of cytotoxicity [14].
2.5. Cell treatment and cytotoxicity assay
All materials were sterilized under UV irradiation for 3 h before their application in tissue culture. Serial dilutions were prepared in 2% RPMI 1640 giving concentrations 100, 50, 25, 12 and 6 μg ml−1. Cytotoxicity was investigated through measurement of cell viability using sulforhodamine B (SRB) assay [15]. Positive and negative cytotoxicity controls were run in each plate. Negative controls (cells with media only; untreated cells) were set as 100% viable. Cells subjected to osmotic shock (treated with distilled water) were taken as positive controls (zero viability) and were used to subtract background from all OD values. The morphological changes of the cells were monitored by phase contrast microscopy (40× magnification).
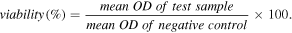
2.6. Imaging and cellular uptake of CS-NPs
Cells were treated with 100 and 1000 μM CS-NPs for 24 h. The cells were washed with phosphate-buffered saline (PBS) and then fixed with 2% glutaraldehyde for 2 h and washed twice with PBS before fixation in 1% OsO4 for 1 h. Following agarose (1.5%) enrobing, Spurr's resin embedding, and ultrathin (50 nm) sectioning, the samples were stained with 2% aqueous uranyl acetate and 25 mg ml−1 lead citrate and imaged with a JEOL 100S microscope [16].
2.7. Flow cytometric cell cycle analysis
HepG2 cells (5 × 105 cells/well) were plated in six-well microplates. After treatment with the IC50 concentration of the CS-NPs, the cells were washed twice with PBS, suspended in 300 μl of PBS, and finally fixed with 4 ml of ice-cold 70% ethanol. To stain them with propidium iodide (PI), cell sedimentation was performed by centrifugation, the ethanol was removed and the cells were washed once with PBS. The cell pellets were then resuspended in 1 ml of PI/Triton X-100 staining solution consisting of 0.1% Triton X-100 in PBS, 0.2 mg ml−1 ribonuclease enzyme A and 10 mg ml−1 PI, and incubated for 30 min at room temperature. The stained cells were analyzed using a MoFlo flow cytometer (DakoCytomation, Glostrup) to calculate the percentages of cells occupying the different phases of the cell cycle [17].
2.8. DNA fragmentation
Fragmentation of cellular DNA was investigated following treatment of the HepG2 cells with CS-NPs at low and high concentrations (100 μM and 1000 μM, IC50, respectively) compared to the estimated IC50. But DNA fragmentation was also investigated following treatment of the HepG2 cells with CS-NPs at low and high concentrations (10 μM and 100 μM, IC50, respectively) compared to the estimated IC50. A fixed amount (100 ng) of cellular DNA (Genomic DNA Purification Kit, Amersham Biosciences) extracted from treated and untreated cells was subjected to 1.5% agarose gel electrophoresis in Tris–acetate buffer pH 8.2, and stained with 0.5 μg ml−1 ethidium bromide. The bands were examined under UV transillumination and photographed. Smearing, or the presence of many low molecular weight DNA fragments, is a characteristic feature of apoptotic cells [18].
2.9. Detection of cellular apoptotic gene expression by one-step reverse-transcription polymerase chain reaction
Extensive evaluation of the cytotoxic effect of CS-NPs on the expression of apoptotic genes (p53, Bak, and caspase 3) at the transcriptional level using one-step reverse-transcription polymerase chain reaction (RT-PCR) assay was performed after treatment of HepG2 with CS-NPs at concentrations of 100 μg ml−1 (high) and 30 μg ml−1 (low) for 24 h and 48 h. The β-actin housekeeping gene was detected in each run to ensure RNA integrity. The detection of mRNA was previously optimized at different annealing temperatures [14, 19].
3. Results
3.1. Synthesis and FTIR analysis of CS-NPs
Nanoparticles were made from CS by the ion gelation method (figure 1). FTIR analysis was required to predict the formation mechanism of the CS-NPs. Also, determination of particle size, charge density and morphology was required, as they are essential characteristic parameters for CS-NPs.

Figure 1. Schematic illustration of the ionotropic gelation process.
Download figure:
Standard image High-resolution imageThe FTIR spectra and the characteristic peaks of the CS, pentasodium TPP, and CS–TPP nanoparticles are shown in table 1 and figure 2.
Table 1. The main characteristic peaks of the chitosan (CS), pentasodium tripolyphosphate (TPP) and chitosan–tripolyphosphate (CS–TPP) nanoparticles.
| Material | Wavenumber (cm−1) | Assignment |
|---|---|---|
| CS | 3360 | N−H stretching vibration overlapped with −OH stretching vibration. |
| 2870 | C−H stretching vibration mode. | |
| 1650 | C=O of amide group –CONH. | |
| 1580 | Bending vibration of N−H. | |
| 1430 | Bending vibration of O−H deformation. | |
| 1370 | Bending vibration of C−H group. | |
| 1080 and 1030 | C−O stretching vibration. | |
| TPP | 1210 | Stretching vibration of P=O. |
| 1130 | Symmetric and anti-symmetric stretching vibration of O−P=O. | |
| 1090 | Symmetric and anti-symmetric stretching vibration of PO3. | |
| 888 | Stretching vibration of P−O−P bridge. | |
| CS–TPP nanoparticles | 3360 broad | N−H stretching vibration overlapped with −OH stretching vibration. |
| 1530 | N−O−P stretching vibration. | |
| 1630 | Bending vibration of N−H. |

Figure 2. (a) The FTIR spectra of pentasodium tripolyphosphate (TPP), chitosan (CS) and chitosan nanoparticles (CS-NPs) in the range 400–4000 cm−1; (b) schematic illustration of the mechanism of chitosan nanoparticle formation.
Download figure:
Standard image High-resolution imageThe spectrum of CS shows the characteristic absorption band at 3360 cm−1 is assigned to the stretching vibration mode of N−H overlapped with the O–H stretching vibration mode. The peak at 2870 cm−1 is attributed to the C−H stretching vibration mode while the peak at 1650 cm−1 is assigned to C=O. The bending vibration of the N−H group appears at 1580 cm−1 whereas the bending vibration of O−H comes out at 1430 cm−1. The peak at 1370 cm−1 is attributed to the bending vibration of C−H deformation. The two peaks at 1080 cm−1 and 1030 cm−1 are assigned to the skeletal vibrations involving O−C stretching. The spectrum of TPP shows the characteristic band at 1210 cm−1 which is assigned to the stretching vibration of P=O. The peak at 1130 cm−1 is assigned to the symmetric and anti-symmetric stretching vibrations in the O−P=O group. The band at 1090 cm−1 is assigned to the symmetric and anti-symmetric stretching vibrations of the PO3 group and the peak at 888 cm−1 can be assigned to the asymmetric stretching vibration of the P−O−P bridge [10–12].
3.2. Characterization of CS-NPs
CS-NPs were characterized in terms of surface morphology, zeta potential, and particle size. As shown in figures 3(a) and (b), CS-NPs are spherical with monodispersity, and CS-NPs are positively charged. The zeta size of CS was determined to be 210 nm (figure 3(c)). The zeta potential was determined by electrophoretic light scattering studies performed in an aqueous solution and the measurement was 40 mV (figure 3(d)).

Figure 3. TEM image of chitosan nanoparticles at scale bar (a) 500 nm and (b) 200 nm, (c) zeta size of chitosan nanoparticles and (d) zeta potential of chitosan nanoparticles.
Download figure:
Standard image High-resolution image3.3. Interaction of CS-NPs with HepG2 cells
3.3.1. Light microscopy
HepG2 cells treated with 100 μM CS-NPs for 24 h did not show profound morphological changes and hence did not reveal any characteristic of cytotoxicity (figure 4).

Figure 4. Treated HepG2 with 100 μg ml−1 of chitosan nanoparticles: (a) morphology of HepG2 and (b) untreated HepG2.
Download figure:
Standard image High-resolution image3.3.2. TEM
TEM images demonstrated the binding and internalization of CS-NPs into HepG2 cells. The aggregation of CS-NPs to form nanoparticle clusters on the cell membrane is evident (figure 5). Examination of the images at higher magnification shows intracellular nanoparticle clusters, mainly associated with membranes, with most of the dispersed nanoparticles found in the cytoplasm.

Figure 5. TEM images of localization of CS-NPs in HepG2 cells in different scale bars: (a) 1.5 μm and (b) 1 μm. Indicated: NP (nanoparticle), CM (cell membrane), Cyto (cytoplasm).
Download figure:
Standard image High-resolution image3.3.3. Cytotoxic effect of CS-NPs in HepG2 cells
The cytotoxic effect of various concentrations of CS-NPs (100, 50, 25, 12.5 and 5 μg ml−1) was assessed in HepG2 cell cultures using SRB colorimetric assay at 48 h time intervals. Results showed that 100 μg ml−1 killed 12% of the cells after 48 h of cell exposure (figure 6), and IC50 reached 239 μg ml−1.

Figure 6. Effect of different concentrations of chitosan nanoparticles (5–100 μg ml−1) on the growth of HepG2 cells as measured by SRB assay.
Download figure:
Standard image High-resolution image3.4. Effect of CS-NPs on cell cycle analysis
The CS-NPs we prepared were further investigated by flow cytometric analysis of the cell cycle and the DNA content of the cells treated with 100 μg ml−1 CS-NPs. Untreated cells showed the expected cell cycle pattern for continuously growing cells, whereas treated cells showed a slight accumulation in the G2/M phase (table 2 and figure 7).
Table 2. Values of chitosan without and with treated HepG2 at different samples.
| Sample | G0/G1 | G2/M | G2/G1 | S | Diploid |
|---|---|---|---|---|---|
| Control | 70.85% | 9.72% | 1.94% | 19.70% | 4.26% |
| Chitosan-treated cells with 100 μg ml−1 CS-NPs | 69.02% | 11.04% | 1.94% | 19.93% | 3.73% |

Figure 7. Cell cycle analysis of chitosan nanoparticles on HepG2: (a) treated HepG2 with 100 μg ml−1 CS-NPs and (b) untreated HepG2.
Download figure:
Standard image High-resolution image3.5. DNA fragmentation of CS-NPs
DNA fragmentation analysis was carried out to investigate the toxic effects of CS-NPs on cell stability and DNA replication. DNA fragmentation characteristics of late apoptosis were observed after treatment of cells with 100 μg ml−1 CS-NPs after 48 h of cell exposure, but such an effect was not observed after 24 h of cell exposure (figure 8, table 3).

Figure 8. EB-stained gel electrophoresis of DNA extraction from untreated and treated HepG2 cell line with CS-NPs: lane 1 (100 bp ladder), lane 2 (CS-NPs 10 μg ml−1), lane 3 (treated HepG2 with 100 μg ml−1 CS-NPs after 24 h), lane 4 (treated HepG2 with 100 μg ml−1 CS-NPs after 48 h) and lane 5 (untreated HepG2; cell control).
Download figure:
Standard image High-resolution imageTable 3. DNA content in DNA fragmentation assay after treatment with chitosan nanoparticles (CS-NPs).
| Sample | Concentration of DNA (ng μl−1) | Ratio (260/280) |
|---|---|---|
| CS-NPs (30 μg ml−1) | 129 | 1.92 |
| CS-NPs (100 μg ml−1) after 24 h | 110 | 1.8 |
| CS-NPs (100 μg ml−1) after 48 h | 30 | 1.7 |
| untreated HepG2 | 139 | 2.2 |
3.6. Apoptotic gene expression as detected by one-step RT-PCR
Our results showed that all selected genes were expressed in the untreated HepG2 cells but the cells treated with 100 μg ml−1 of CS-NPs showed the expression of p53, Bak, and caspase 3 after 24 h and of p53 and Bak after 48 h but did not show the expression of caspase 3 after 48 h. The results are shown in figure 9. All studied apoptotic genes were normally expressed in the cells treated with a low concentration of CS-NPs (30 μg ml−1) after 24 and 48 h of cell exposure.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. EB-stained gel electrophoresis of genomic caspase (Cas) 3, β-actin, Bak and p53 RNA expression from HepG2 treated with CS-NPs at low (30 μg l−1) and high (100 μg ml−1) concentrations after 24 h and 48 h of cell exposure. Lanes 1–4: caspase mRNA expression; lane 5: 50 bp ladder; lanes 6–9: β-actin mRNA gene expression; lanes 10–13: Bak mRNA expression; lanes 14–15: p53 mRNA gene expression from HepG2 treated with high and low CS-NPs after 48 h of cell exposure.
Download figure:
Standard image High-resolution image{kind=link}
4. Discussion
CS-NPs are inexpensive and thus are among the most extensively studied nanoparticles as potential drug carriers with great efficiency in controlled drug release [2]. On the other hand, it has been reported that soluble CS and CS microspheres show some degree of toxicity towards certain cell lines like the murine melanoma cell line and human gastric carcinoma MGC803 cell line suggesting their application as antitumor drugs [20]. Therefore, in the present study we developed our own soluble CS-NPs and evaluated their toxic effect against human liver cancer cells to explore their possible application as a drug carrier system for anti-cancerous and antiviral drugs.
Our freshly prepared CS-NPs were controlled to a size of 150 nm and surface charges of 40 mM. It has been reported that nanoparticles with a zeta potential above ±30 mV are stable in suspension, as the surface charge prevents the aggregation of the particles [21]. Moreover, the absorption peak of CS–TPP nanoparticles at 3360 cm−1 indicates that hydrogen bonding is enhanced. The 1580 cm−1 peak of –NH bending vibration shifts to 1630 cm−1 and a new peak appears at position 1530 cm−1 which is assigned to N-O-P stretching vibration. This indicates that the TPP anions were cross-linked with the ammonium groups of CS to form CS-NPs as illustrated in figure 2. Similar results were also obtained in other works [11, 12].
In the present study, HepG2 was used as a model for human liver cancer. Our results showed that treatment of HepG2 with CS-NPs at 100 μg ml−1 did not reveal any morphological alteration after 24 h and 48 h. The results of our cytotoxicity studies by SRB colorimetric assay [22] showed that treatment of HepG2 with 100 μg ml−1 CS-NPs kills only 12% of the cells after 48 h of cell exposure. Moreover, the inhibition rate increased with increasing CS-NP concentration in the culture medium, where IC50 was found to reach 230 μg ml−1. This indicates that the inhibition of cell viability by CS-NPs was clearly dose- and time-dependent. It has been reported that several parameters affect the cytotoxic profile of CS-NPs like their size, surface properties, degree of deacetylation (46%–88%), and degree of solidification by different amounts of glutaraldehyde and concentrations of CS-NPs [23]. Regarding the extent of solidification of CS-NPs, it has been reported that CS-NPs with a greater extent of solidification exhibit a more vigorous inhibitory effect against HepG2 cells than those without solidification even when treating cells with a low concentration of CS-NPs (18 μg ml−1) [4]. Moreover, it has been observed that CS-NPs prepared from CS of a molecular weight of 10–213 kDa at a size of 110–390 nm and a degree of deacetylation of 46%–88% show comparable cytotoxicity profiles against A549 (human lung adenocarcinoma) cells with cell viability generally not affected by sample concentrations lower than 740 μg ml−1 [24]. In contrast, this was different when using the MGC803 human gastric carcinoma cell line where the IC50 value was 16.2 and 5.3 μg ml−1 after 24 h and 48 h, respectively, in incubation with CS with a size of 65 nm and high charges of 51 mV [20]. This indicates that small size, high positive charges and the type of biologically interacting cells and cellular uptake dramatically affect the cytotoxic effect of CS-NPs.
Hepatic cells also showed divergent responses to the physicochemical properties of CS-NPs. HepG2 cells were found to be unaffected by galactosylated CS-NPs 108 ± 23 nm at concentrations of up to 25 μg ml−1 [25]. IC50 values of 15.01 and 6.19 μg ml−1 were obtained for another human HCC-derived cell line, the BEL7402 cells, after 24 h and 48 h of incubation, respectively, with CS-NPs of 40 nm [26]. In the present study, our CS-NPs at a size of 150 nm and charges of 45 mV showed only a 12% toxic effect on HepG2 at a concentration of 100 μg ml−1 after 48 h of cell exposure.
The cellular uptake and transport of CS-NPs into human liver cells have been reported [23]. It has been observed that CS-NP uptake and cytoadhesion are well correlated to the zeta potential and size of the CS-NPs and CS molecules in situ. These are in agreement with our findings which revealed the internalization of CS-NPs into cytoplasm and nuclei. The mechanism of nanoparticle uptake in hepatocytes is unknown, although it could be similar to the clathrin-mediated endocytic pathway reported for the uptake of CS-NPs in human intestinal cells [27].
Flow cytometric analysis and cellular DNA fragmentation provide insight into the nature of cell death exerted by CS-NPs. Our results showed that treatment of cells with 100 μg ml−1 CS-NPs slightly arrested cells in the G2/M phase but did not reveal much effect on DNA. But by increasing the time of exposure to 48 h, it exerted a dramatic effect on cellular DNA concentration when compared to the untreated cells or even to those treated for shorter time (24 h). Moreover, extensive evaluation of the cytotoxic effect of CS-NPs at high concentration (100 μg ml−1) after extending exposure time to 48 h was conducted for the expression of some apoptotic genes like p53, caspase 3 and Bak on the RNA level. Results showed that the mRNA of β-actin, p53, Bak, and caspase 3 was expressed after 24 h and after 48 h except genomic caspase 3 mRNA. This may indicate that the apoptotic effect of CS-NPs was exerted via a caspase-independent pathway but such results need to be confirmed by quantitative assay and on the protein level.
5. Conclusion
In the present study, CS-NPs 150 nm in diameter at a concentration of 100 μg ml−1 did not show much toxic effect (12%) on human liver cancer cells (hepG2) after 48 h of cell exposure. Such results were followed up by flow cytometry and cellular DNA fragmentation which showed the accumulation of cells in the G2/M phase and a dramatic effect on DNA concentration after 48 h of cell exposure suggesting the possible application of CS-NPs as antitumor drugs against human liver cancer cells; however, this requires further verifications. Therefore, delivery strategies using CS-NPs as a vehicle need to consider their adverse effects upon extending the time of exposure. Therefore, we suggest using our CS-NPs at a concentration of 100 μg ml−1 to encapsulate antitumor drugs against liver cancer cells but decreasing this to 30 μg ml−1 when encapsulating antiviral drugs against HCV replication. Further analysis is still required to investigate different concentrations of smaller sizes of CS-NPs.
Acknowledgments
Our thanks to Dr Zainab Fathy, Virology and Immunology Unit, Cancer Biology Department, National Cancer Institute, Cairo, for establishing the protocol of apoptotic mRNA gene expression. Also we thank Dr Samir Amer, National Cancer Institute, for his efforts in analyzing the flow cytometry section.