

Abstract
Loading of anticancer drugs into electrospun fiber matrices is a portentous approach for clinical treatment of diseased tissues or organs. In this study, doxorubicin hydrochloride (DOX) is added to silica nanoparticles ( ) during the formation of
) during the formation of  via the sol-gel approach. The obtained
via the sol-gel approach. The obtained  nanoparticles are then added to poly(
nanoparticles are then added to poly( -caprolactone) (PCL) and poly(ethylene oxide) (PEO) blend before electrospinning process via different methods. The effects of DOX addition as a free form or as
-caprolactone) (PCL) and poly(ethylene oxide) (PEO) blend before electrospinning process via different methods. The effects of DOX addition as a free form or as  nanoparticles on physical and chemical properties of obtained PCL-PEO fibers, as well as release profiles are evaluated to give a continual DOX release for several days. The morphology observed with scanning electron microscope (FESEM) revealed significant changes in the average diameter of obtained fibers ranging from 2164 nm to 659 nm and distribution of drug-loaded nanoparticles in the final mats according to the mode of additions. With the same manner, the releasing performances of obtained mats are quite different. Therefore, fabrication of drug loaded mats would offer a powerful approach to minimize serious side effects for clinical patients and allows us to control the drug concentration in the bloodstream.
nanoparticles on physical and chemical properties of obtained PCL-PEO fibers, as well as release profiles are evaluated to give a continual DOX release for several days. The morphology observed with scanning electron microscope (FESEM) revealed significant changes in the average diameter of obtained fibers ranging from 2164 nm to 659 nm and distribution of drug-loaded nanoparticles in the final mats according to the mode of additions. With the same manner, the releasing performances of obtained mats are quite different. Therefore, fabrication of drug loaded mats would offer a powerful approach to minimize serious side effects for clinical patients and allows us to control the drug concentration in the bloodstream.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Numerous drugs have inadmissible side effects owing to the drug interaction with healthy organs or tissues that are not the target of the drug. The unwanted drugs side effects limit our capability to formulate the optimal medications for clinical curing of various diseases such as cancer, neurodegenerative and some infectious diseases [1, 2]. Recent progress in regenerative medicine and biomaterials lead to improve drug administration method via decreasing free drug distribution within the body and targeting diseased sites. In this context, drug delivery systems are designed to exhibit the potential of sustain and target delivery of drugs, therefore allow for improved efficacy of chemotherapy, reduced painful side effects and decreased frequency of drug administration [3, 4].
Natural or synthetic biodegradable polymers have a set of priorities in the biomedical area as scaffold matrices used for tissue engineering applications and as vesicles for drug delivery vehicles to concentrate specific bioactive molecules such as drugs or growth factors in the diseased tissue of interest or to target a definite organelle through the cells [5]. Electrospun polymeric micro- or nanofibers have been applied as a drug carrier, because of the wide scope for direct incorporation of hydrophobic and hydrophilic drugs also biomacromolecules like proteins or RNA into the electrospun fibers mats [6]. The interconnected porous feature and high surface area of electrospun fibers help us to decrease the drug dose within the implant, leading to a less systemic toxicity and reduce not desired side effects. Moreover, the release profile of drug from the electrospun webs can be modified by adjusting the fiber morphology, porosity and texture [7]. Therefore, electrospun polymer fibers would provide an ersatz approach for long-term antitumor activity via incorporation of the target therapeutic agent into electrospun fibers [4]. Several anticancer drugs have been loaded into various electrospun fibers for postsurgical cancer therapy [8, 9]. Doxorubicin hydrochloride (DOX) is one of the most widely employed chemotherapeutic drugs in cancer treatment. It is a class of anthracycline antibiotic which acts on the S-phase of the cell cycle and causes cell death by damaging DNA and its synthesis through the intercalation between nucleotides, generation of oxygen-free radicals and inhibition of topoisomerase II [10]. When the drug loaded into the surface of the nanofibers an undesirable burst release is constantly inevitable in account of their high ionic strength in solution and the solvent quick evaporation through electrospinning [11]. To overcome this issue, several inorganic nanocarriers have been entrapped into electrospun nanofibers so as to prevent initially burst release of drug and provide a slow sustainable release [12]. Silica nanoparticles are chosen as the drug carrier as: silica nanoparticles (SiO2), have raised much interest in the preparation of drug delivery systems because of their high surface areas and porous interiors which permit them to be used as reservoirs for loading both hydrophilic and hydrophobic drugs. In addition, SiO2 was reported to be able to promote the dissolution of the indigent water-soluble indications and enhance their bioavailability [2, 13, 14].
Among the famous synthetic polymers used in electrospining are hydrophobic polycaprolactone (PCL) and hydrophilic polyethylene oxide (PEO). PCL is a semi-crystalline biodegradable thermoplastic polymer and has been confirmed by the Food and Drug Administration (FDA) in the USA as a biodegradable material for biomedical purposes as drug delivery and tissue engineering applications [15]. PEO is a water-soluble amphiphilic polymer with good biological features, including biocompatibility and soft toxicity [16]. This work aimed to load DOX (as an anticancer drug) into the silica nanoparticles and find out the right set of conditions to incorporate drug loaded nanoparticles into PCL-PEO nanofiber mats. The chemical also physical characteristics of the fabricated fiber mats and nanoparticles are characterized. In addition, the suitability of the prepared polymeric mats, as a substrate for drug release application, was investigated by studying the drug releasing profile.
2. Materials and methods
2.1. Materials
Poly( -caprolactone) (PCL with average molecular weight
-caprolactone) (PCL with average molecular weight  ) and polyethylene oxide (PEO) with molecular weight
) and polyethylene oxide (PEO) with molecular weight  ) were bought from Sigma Aldrich. Doxorubicin hydrochloride (DOX, Mw = 580) was secured from the Beijing Huafeng United Technology Co, Ltd. Chloroform purchased and tetraethyl orthosilicate (TEOS) were get from Sigma-Aldrich. Methanol, absolute ethanol and ammonia solution (25%) were obtained from El-Nasr Company.
) were bought from Sigma Aldrich. Doxorubicin hydrochloride (DOX, Mw = 580) was secured from the Beijing Huafeng United Technology Co, Ltd. Chloroform purchased and tetraethyl orthosilicate (TEOS) were get from Sigma-Aldrich. Methanol, absolute ethanol and ammonia solution (25%) were obtained from El-Nasr Company.
2.2. Methods
2.2.1. Preparation of  and .
and .
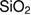
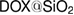
Silica nanoparticles are prepared via sol-gel process based on the Stöber method using tetraethyl orthosilicate (TEOS,  ) as a precursor [17]. In brief,
) as a precursor [17]. In brief,  ethanol and
ethanol and  ammonia solution (25%) is stirred together for about 30 min in a closed polyethylene bottle. Then,
ammonia solution (25%) is stirred together for about 30 min in a closed polyethylene bottle. Then,  TEOS is appended to the above solution and is further stirred at room temperature for 24 h. The particles in the sol are afterward gathered by centrifugation and washed three times with distilled water. Finally, the obtained wet particles are dried at 40 °C for about 24 h. For the drug loaded silica nanoparticles, DOX is incorporated during the formation of silica nanoparticles. At first, different amounts of DOX with respect to weight of silica (0.5, 2, 4%wt of
TEOS is appended to the above solution and is further stirred at room temperature for 24 h. The particles in the sol are afterward gathered by centrifugation and washed three times with distilled water. Finally, the obtained wet particles are dried at 40 °C for about 24 h. For the drug loaded silica nanoparticles, DOX is incorporated during the formation of silica nanoparticles. At first, different amounts of DOX with respect to weight of silica (0.5, 2, 4%wt of  ) are dissolved in ethanol/ammonia solution before TEOS addition. The obtained
) are dissolved in ethanol/ammonia solution before TEOS addition. The obtained  nanoparticles are named
nanoparticles are named  ,
,  and
and  , respectively.
, respectively.
2.2.2. Preparation of the electrospinning solution.
PCL with an optimized concentration (10%) and PEO with an optimized concentration (3% relative to PCL) are dissolved in a blend of chloroform:methanol (3:1 v/v) for all the samples. The PCL-PEO solution is loaded into a 1 ml plastic syringe to which a metal blunt ended needle (G18) is attached. The injection velocity through the syringe pump into the nozzle is adjusted with a flow rate at  . The high voltage supply positive output lead is attached to the needle on the syringe, while the working platform is connected with the negative pole. For drug contained fiber, five methods are applied to incorporate DOX and
. The high voltage supply positive output lead is attached to the needle on the syringe, while the working platform is connected with the negative pole. For drug contained fiber, five methods are applied to incorporate DOX and  nanoparticles into the polymer solution as follow: (i) method (named
nanoparticles into the polymer solution as follow: (i) method (named  ), a definite amount of DOX are added to PCL-PEO solution. After dissolution, the prepared solution is transferred to a
), a definite amount of DOX are added to PCL-PEO solution. After dissolution, the prepared solution is transferred to a  plastic syringe for electrospinning. (ii) Method (named
plastic syringe for electrospinning. (ii) Method (named  ), DOX loaded to
), DOX loaded to  nanoparticles are mixed with the polymer solution instead of free DOX. (iii) Method (named
nanoparticles are mixed with the polymer solution instead of free DOX. (iii) Method (named  ), the PCL-PEO and
), the PCL-PEO and  mixture is sonicated for about 30 min. (iv) Method (named
mixture is sonicated for about 30 min. (iv) Method (named  ), the
), the  nanoparticles are separately sonicated for about 30 min before the addition to PCL-PEO solution. The obtained mixture are again stirred for 1 h and sonicated for 30 min. (v) Method (named
nanoparticles are separately sonicated for about 30 min before the addition to PCL-PEO solution. The obtained mixture are again stirred for 1 h and sonicated for 30 min. (v) Method (named  ) (core shell),
) (core shell),  nanoparticles are dispersed in chloroform/methanol mixture via ultrasonic and then loaded into
nanoparticles are dispersed in chloroform/methanol mixture via ultrasonic and then loaded into  plastic syringe. The PCL-PEO solution is transferred into another syringe fitted with a needle having a tip diameter larger than the tip diameter of the syringe used for
plastic syringe. The PCL-PEO solution is transferred into another syringe fitted with a needle having a tip diameter larger than the tip diameter of the syringe used for  nanoparticles solution. Finally, the formed electrospun mats are dried at room temperature for 24 h to permit the total solvent removal.
nanoparticles solution. Finally, the formed electrospun mats are dried at room temperature for 24 h to permit the total solvent removal.
2.3. Characterization
2.3.1. Microstructure characterizations.
The morphology of the electrospun fibers is examined under FESEM that processed using FESEM model (Quanta FEG 250 FEI, Holanda). The diameter of the fiber and their distribution is estimated from image j software. The as prepared silica and  nanoparticles are examined by TEM, JeolJem-1230. Fourier transform infrared spectroscopy (FTIR) of all mats is performed by Jasco, FTIR—6100 type A. The FTIR spectra are obtained in the region
nanoparticles are examined by TEM, JeolJem-1230. Fourier transform infrared spectroscopy (FTIR) of all mats is performed by Jasco, FTIR—6100 type A. The FTIR spectra are obtained in the region  for all samples. Phase analysis of the prepared samples is applied using x-ray diffractrometer Bruker D8 advance CuK target with secondary monochromator at
for all samples. Phase analysis of the prepared samples is applied using x-ray diffractrometer Bruker D8 advance CuK target with secondary monochromator at  and
and  . The thermal properties and weight changes are analyzed as a function of temperature by thermal gravimetric analysis (TGA), NETZSCH STA 409 instrument. The test is performed at a heating rate of
. The thermal properties and weight changes are analyzed as a function of temperature by thermal gravimetric analysis (TGA), NETZSCH STA 409 instrument. The test is performed at a heating rate of  under nitrogen atmosphere from RT up to
under nitrogen atmosphere from RT up to  . Differential scanning calorimetry (DSC) measurements are carried out using Mettler DSC 822/400) thermal analyzer instrument having sub-ambient capability.
. Differential scanning calorimetry (DSC) measurements are carried out using Mettler DSC 822/400) thermal analyzer instrument having sub-ambient capability.
2.3.2. In vitro drug release.
All the electrospun mats are cut into  square pieces. The sample weight is recorded before dipping into a centrifuge tube filled with 10 ml of phosphate buffered saline solution (PBS). At time periods of 1 h, 2 h, 3 h, 1 d, 2 d, 6 d, 10 d, 14 d, 21 d and 33 d the amount of DOX released in the PBS is measured using UV-Vis spectrophotometer by measuring the absorbance at 480 nm. The experiments are processed in triplicate per sample. The percentage of released drug is calculated using the following equation:
square pieces. The sample weight is recorded before dipping into a centrifuge tube filled with 10 ml of phosphate buffered saline solution (PBS). At time periods of 1 h, 2 h, 3 h, 1 d, 2 d, 6 d, 10 d, 14 d, 21 d and 33 d the amount of DOX released in the PBS is measured using UV-Vis spectrophotometer by measuring the absorbance at 480 nm. The experiments are processed in triplicate per sample. The percentage of released drug is calculated using the following equation:

where  is the mass of DOX released at time
is the mass of DOX released at time  and
and  is the total amount of DOX present on the fibrous mat [4].
is the total amount of DOX present on the fibrous mat [4].
3. Results and discussion
3.1. Morphological observations of electrospun PCL/PEO fibers
Figure 1 shows the FESEM images of the electrospun PCL-PEO fibers loaded with DOX and  via different methods at different magnifications. As can be seen in figures 1(A), (A1) and (A2),
via different methods at different magnifications. As can be seen in figures 1(A), (A1) and (A2),  method leads to agglomeration of DOX particles on the surface of the fibers. This could be due to the diameter of PCL-PEO fiber which is smaller than DOX aggregates. The average diameter of PCL-PEO fiber is
method leads to agglomeration of DOX particles on the surface of the fibers. This could be due to the diameter of PCL-PEO fiber which is smaller than DOX aggregates. The average diameter of PCL-PEO fiber is  (figure 2(M1)). The same agglomeration is also seen for
(figure 2(M1)). The same agglomeration is also seen for  nanoparticles loaded on the fibers by the M2 method as shown in (figures 1(B), (B1) and (B2)) with an average diameter of about
nanoparticles loaded on the fibers by the M2 method as shown in (figures 1(B), (B1) and (B2)) with an average diameter of about  (figure 2(M2)). On the other hand, the obtained
(figure 2(M2)). On the other hand, the obtained  method fiber shows less agglomeration of
method fiber shows less agglomeration of  . The dumbbell-shaped features of PCL-PEO suggest the encapsulation of drug particles inside the nanofiber shown in (figures 1(C), (C1) and (C2)). This could be due to utrasonication of the samples before electrospining. The average diameter of the obtained fibers is 1492.124 nm (figure 2(M3)). However, the loading of
. The dumbbell-shaped features of PCL-PEO suggest the encapsulation of drug particles inside the nanofiber shown in (figures 1(C), (C1) and (C2)). This could be due to utrasonication of the samples before electrospining. The average diameter of the obtained fibers is 1492.124 nm (figure 2(M3)). However, the loading of  by the
by the  and
and  method resulted to the insertion of the all particles inside the electrospun PCL-PEO nanofibers as shown in (figures 1(D), (D1), (D2), (E), (E2) and (E3)). The average diameter of obtained fibers is increased to 1880.713 nm for
method resulted to the insertion of the all particles inside the electrospun PCL-PEO nanofibers as shown in (figures 1(D), (D1), (D2), (E), (E2) and (E3)). The average diameter of obtained fibers is increased to 1880.713 nm for  fibers and
fibers and  for
for  method (figures 2(M4) and (M5)). Some of the
method (figures 2(M4) and (M5)). Some of the  nanoparticles find in agglomeration inside the fibers caused the formation of the swollen beads on the nanofibers as shown by the dashed green circles. The above results focus on the effect of ultrasonic on morphology and nanoparticles distribution within the fiber mats, in which the loaded nanoparticles are highly agglomerate in absence of ultrasonic (
nanoparticles find in agglomeration inside the fibers caused the formation of the swollen beads on the nanofibers as shown by the dashed green circles. The above results focus on the effect of ultrasonic on morphology and nanoparticles distribution within the fiber mats, in which the loaded nanoparticles are highly agglomerate in absence of ultrasonic ( ) and less agglomeration distribution, is obtained with
) and less agglomeration distribution, is obtained with  due to the sonication that separate agglomerated particles from each other. In this consequence, uniform distribution is achieved via sonication of
due to the sonication that separate agglomerated particles from each other. In this consequence, uniform distribution is achieved via sonication of  nanoparticles before and after the addition to the polymer solution,
nanoparticles before and after the addition to the polymer solution,  method. Finally,
method. Finally,  method provides an alternative approach for high drug encapsulation capability as presented by FESEM micrographs.
method provides an alternative approach for high drug encapsulation capability as presented by FESEM micrographs.

Figure 1. FESEM micrographs of electrospun PCL-PEO loaded with DOX and  nanoparticles via different methods at diverse magnifications: (A), (A1) and (A2) DOX loaded by M1 method; (B), (B1) and (B2)
nanoparticles via different methods at diverse magnifications: (A), (A1) and (A2) DOX loaded by M1 method; (B), (B1) and (B2)  loaded by M2 method; (C), (C1) and (C2)
loaded by M2 method; (C), (C1) and (C2)  loaded by M3 method, (D), (D1) and (D2)
loaded by M3 method, (D), (D1) and (D2)  loaded by M4 method and (E), (E1) and (E2)
loaded by M4 method and (E), (E1) and (E2)  loaded by M5 method. Red circle points to the existence of agglomerated DOX (A1) or
loaded by M5 method. Red circle points to the existence of agglomerated DOX (A1) or  (B1) and (C1) on the surface of fibers, while dashed green circle (C1), (D1) and (E1) points to the encapsulation of nanoparticles inside the fibers.
(B1) and (C1) on the surface of fibers, while dashed green circle (C1), (D1) and (E1) points to the encapsulation of nanoparticles inside the fibers.
Download figure:
Standard image High-resolution image
Figure 2. The distributions and average diameters of electrospun PCL-PEO fibers via different methods of preparation.
Download figure:
Standard image High-resolution image3.2. The microstructure analysis of PCL-PEO fibers and nanoparticles
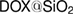
The chemical structure analyses of the obtained fiber mats and  nanoparticles are characterized by FTIR (figure 3). The FTIR spectrum of pure PCL-PEO nanofiber is shown in curve A of figure 3(a) through this spectrum the band at about,
nanoparticles are characterized by FTIR (figure 3). The FTIR spectrum of pure PCL-PEO nanofiber is shown in curve A of figure 3(a) through this spectrum the band at about,  is associated with CO − stretching vibration of ester in PCL, the band occurred at
is associated with CO − stretching vibration of ester in PCL, the band occurred at  is related to the O–C–O asymmetric stretching vibrations of PCL, and that of symmetric stretching occur at
is related to the O–C–O asymmetric stretching vibrations of PCL, and that of symmetric stretching occur at  , the bands at about
, the bands at about  and
and  may be associated with the vibration of C–O present on PCL and the vibrational band at about
may be associated with the vibration of C–O present on PCL and the vibrational band at about  is related to
is related to  of PCL [18].
of PCL [18].

Figure 3. (a) FTIR spectra of pure PCL-PEO fibers (curve A) and of different fibrous mats (curves B, C, D, E and F) prepared by different methods ( and
and  , respectively); and (b) FTIR spectra of pure SiO2 nanoparticles and
, respectively); and (b) FTIR spectra of pure SiO2 nanoparticles and  and
and  .
.
Download figure:
Standard image High-resolution imageThe intense doublet band at about  and
and  assigned to asymmetric and symmetric vibration of
assigned to asymmetric and symmetric vibration of  of PEO, respectively. The two bands at
of PEO, respectively. The two bands at  and
and  are concerned with scissoring and twisting vibration of
are concerned with scissoring and twisting vibration of  , respectively [19]. The band at
, respectively [19]. The band at  is associated with wagging vibration of
is associated with wagging vibration of  belongs to PCL and PEO. Also the band at about
belongs to PCL and PEO. Also the band at about  is assigned to rocking vibration of
is assigned to rocking vibration of  found on PCL and PEO. On the other hand, there are some changes occur on the FTIR spectra of PCL-PEO fibers loaded with DOX or
found on PCL and PEO. On the other hand, there are some changes occur on the FTIR spectra of PCL-PEO fibers loaded with DOX or  . As illustrated in curves B, C and D of figure 3(a) for
. As illustrated in curves B, C and D of figure 3(a) for  and
and  methods, respectively, we can see that the bands at about
methods, respectively, we can see that the bands at about  ,
,  ,
,  ,
,  ,
,  do not exist in the spectra. Also the intensity of the band at about
do not exist in the spectra. Also the intensity of the band at about  and intensity of doublet bands at about
and intensity of doublet bands at about  and
and  decrease in these spectra as compared to the spectrum of pure PCL-PEO. All these alterations could be related to the presence or adsorption of loaded particles on their surface. For the other methods
decrease in these spectra as compared to the spectrum of pure PCL-PEO. All these alterations could be related to the presence or adsorption of loaded particles on their surface. For the other methods  and
and  , the spectra are resembled to the spectrum of pure PCL-PEO which may be because of
, the spectra are resembled to the spectrum of pure PCL-PEO which may be because of  particles that are encapsulated by PCL-PEO fibers.
particles that are encapsulated by PCL-PEO fibers.
Figure 3(b) shows the FTIR spectra of pure SiO2 and samples of DOX loaded on silica nanoparticles at different ratios 0.5, 2, 4% named  , respectively). The spectrum of pure
, respectively). The spectrum of pure  nanoparticles illustrates the presence of absorption bands arising from asymmetric vibration of Si–O on the range of (
nanoparticles illustrates the presence of absorption bands arising from asymmetric vibration of Si–O on the range of ( ), asymmetric vibration of Si–OH at (
), asymmetric vibration of Si–OH at ( ), and symmetric vibration of Si–O appeared at (
), and symmetric vibration of Si–O appeared at ( ).The intense characteristic absorption band at the range (
).The intense characteristic absorption band at the range ( ) ascribed to O–H of stretching in H-bonded water. Also the band assigned at (
) ascribed to O–H of stretching in H-bonded water. Also the band assigned at ( ) is due to scissor bending vibration of molecular water. The loading of DOX into the silica nanoparticles led to obvious changes on the vibrational bands. The broadening of asymmetric vibration band of Si–O increased with the loading of drug into the silica nanoparticles. The intensity of vibration band of Si–OH decreases with the incorporation of DOX into the silica nanoparticles and shifts to higher wavenumbers. This may be due to: presence of drug into the silica nanoparticles increases their diameters which in turn decrease the length of the bond between Si–OH which cause its shift to higher wavenumbers.
) is due to scissor bending vibration of molecular water. The loading of DOX into the silica nanoparticles led to obvious changes on the vibrational bands. The broadening of asymmetric vibration band of Si–O increased with the loading of drug into the silica nanoparticles. The intensity of vibration band of Si–OH decreases with the incorporation of DOX into the silica nanoparticles and shifts to higher wavenumbers. This may be due to: presence of drug into the silica nanoparticles increases their diameters which in turn decrease the length of the bond between Si–OH which cause its shift to higher wavenumbers.
TEM images of pure  and
and  nanoparticles are indicated in figures 4(A), (B), (B1) and (B2). As can be seen, the as prepared pure silica nanoparticles are spherical in shape that agglomerated to form clusters. The loading of DOX into the silica nanoparticles leads to formation of uniform spheres larger than pure silica. The diameter of
nanoparticles are indicated in figures 4(A), (B), (B1) and (B2). As can be seen, the as prepared pure silica nanoparticles are spherical in shape that agglomerated to form clusters. The loading of DOX into the silica nanoparticles leads to formation of uniform spheres larger than pure silica. The diameter of  nanoparticles are ranged from
nanoparticles are ranged from  . In addition, figure 4(C) shows the XRD spectra of the synthesized pure silica nanoparticles and DOX loaded silica nanoparticles. The XRD pattern of pure SiO2 exhibits a broad peak at
. In addition, figure 4(C) shows the XRD spectra of the synthesized pure silica nanoparticles and DOX loaded silica nanoparticles. The XRD pattern of pure SiO2 exhibits a broad peak at  , which reveals the amorphous nature of the silica nanoparticles. The broadening of this peak is obvious owing to the smaller grain size effect. The absence of other peaks indicates that there are no impurities in the prepared silica nanoparticles [20]. For
, which reveals the amorphous nature of the silica nanoparticles. The broadening of this peak is obvious owing to the smaller grain size effect. The absence of other peaks indicates that there are no impurities in the prepared silica nanoparticles [20]. For  sample, there is a small shift of
sample, there is a small shift of  to
to  , as indicated from the XRD pattern in figure 4(C). In addition, the broadening of the peak decreases slightly with the loading of DOX into the silica nanoparticles. These changes reflect the incorporation of DOX within the structure of silica nanoparticles.
, as indicated from the XRD pattern in figure 4(C). In addition, the broadening of the peak decreases slightly with the loading of DOX into the silica nanoparticles. These changes reflect the incorporation of DOX within the structure of silica nanoparticles.

Figure 4. TEM images of (A) pure  , and (B), (B1) and (B2) of
, and (B), (B1) and (B2) of  ; (C) XRD patterns of pure
; (C) XRD patterns of pure  and 4%
and 4%  .
.
Download figure:
Standard image High-resolution image3.3. Thermal analysis of and

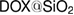
For investigating the amount of encapsulated DOX into the  nanoparticles excluding the interference of the drug content changes into the measurement, the TGA thermal analysis of pure silica nanoparticles and loaded with doxorubicin is investigated. Figures 5(A)–(C) illustrate the stages of weight loss. The TGA analysis of pure silica nanoparticles exhibited four stages of weight loss which could be indicated as follow: first stage of weight loss occurred at around
nanoparticles excluding the interference of the drug content changes into the measurement, the TGA thermal analysis of pure silica nanoparticles and loaded with doxorubicin is investigated. Figures 5(A)–(C) illustrate the stages of weight loss. The TGA analysis of pure silica nanoparticles exhibited four stages of weight loss which could be indicated as follow: first stage of weight loss occurred at around  , in which the mass loss during this stage is about 0.38%. This is related to the vaporization of physically adsorbed water on the silica surface. Second stage of weight loss happens during the temperature range about
, in which the mass loss during this stage is about 0.38%. This is related to the vaporization of physically adsorbed water on the silica surface. Second stage of weight loss happens during the temperature range about  which contributes to a weight loss of about 3.87%. The loss in weight for this stage is related to the escape of entrapped water and apportion of residual solvent (ethanol) in the porous structure of silica matrix. The third stage of weight loss covers the range of temperature (
which contributes to a weight loss of about 3.87%. The loss in weight for this stage is related to the escape of entrapped water and apportion of residual solvent (ethanol) in the porous structure of silica matrix. The third stage of weight loss covers the range of temperature ( ). During this stage the weight loss (2.3%) may be due to the dehydroxylation of the silanol group and the degradation of residual reactant (TEOS). The fourth stage
). During this stage the weight loss (2.3%) may be due to the dehydroxylation of the silanol group and the degradation of residual reactant (TEOS). The fourth stage  is suggested to be due to the degradation of residual TEOS and dehydroxylation of silanol groups. The weight loss at this stage is about 1.44% [21]. Therefore, the total weight loss of silica nanoparticles along all range of temperature
is suggested to be due to the degradation of residual TEOS and dehydroxylation of silanol groups. The weight loss at this stage is about 1.44% [21]. Therefore, the total weight loss of silica nanoparticles along all range of temperature  is about 7.986% of the total mass. This means that the prepared silica nanoparticles are thermally stable up to
is about 7.986% of the total mass. This means that the prepared silica nanoparticles are thermally stable up to  . The observation of TGA thermal analysis of
. The observation of TGA thermal analysis of  shows that there are apparently two stages of weight loss: (i) first one occurred over the temperature range of
shows that there are apparently two stages of weight loss: (i) first one occurred over the temperature range of  , the weight loss in this case is about 53.22%, which may due to loss of water, large portion of solvents and also the decomposition of all present DOX; (ii) second stage appears to cover the temperature range of
, the weight loss in this case is about 53.22%, which may due to loss of water, large portion of solvents and also the decomposition of all present DOX; (ii) second stage appears to cover the temperature range of  , the weight loss is about 8.69%, this may be due to the escape of the residual solvents. The overall amount of loaded DOX is estimated by TGA. In the TGA measurement, the drug desorbs after decomposing, which is detected as a temperature dependent weight reduction. However quantification of the loaded-drug fraction in the pores of material is not as simple as just quantifying the total drug content of the sample [22]. The loading fractions are estimated from the mass loss ratio between
, the weight loss is about 8.69%, this may be due to the escape of the residual solvents. The overall amount of loaded DOX is estimated by TGA. In the TGA measurement, the drug desorbs after decomposing, which is detected as a temperature dependent weight reduction. However quantification of the loaded-drug fraction in the pores of material is not as simple as just quantifying the total drug content of the sample [22]. The loading fractions are estimated from the mass loss ratio between  and
and  compared to the total initial weight. Since the total weight loss of pure silica is about 7.986%, while that for
compared to the total initial weight. Since the total weight loss of pure silica is about 7.986%, while that for  is about 61.91%, therefore the loading of DOX into the silica nanoparticles is about 53.92%.
is about 61.91%, therefore the loading of DOX into the silica nanoparticles is about 53.92%.

Figure 5. TGA indicates stages for weight loss of (A) pure  , (B)
, (B)  and both pure
and both pure  and
and  (C). (D) DSC thermogram of pure SiO2 and
(C). (D) DSC thermogram of pure SiO2 and  .
.
Download figure:
Standard image High-resolution imageFigure 5(D) shows the DSC thermal analysis of the pure silica nanoparticles and  . For pure silica nanoparticles, there is an endothermic peak centered at
. For pure silica nanoparticles, there is an endothermic peak centered at  . This is attributed to removal of physically adsorbed water molecules, ethanol and ammonia at the surface of silica. The endothermic peak at about
. This is attributed to removal of physically adsorbed water molecules, ethanol and ammonia at the surface of silica. The endothermic peak at about  may be due to removal of residual water present on pores and vaporization of other solvents present due to the dehydroxylation process (release of water molecules due to condensation of silanol groups and formation of siloxane linkage) [21–23]. For
may be due to removal of residual water present on pores and vaporization of other solvents present due to the dehydroxylation process (release of water molecules due to condensation of silanol groups and formation of siloxane linkage) [21–23]. For  , the first endothermic peak centered for the pure silica and
, the first endothermic peak centered for the pure silica and  at the same temperature is due to dehydration. Doxorubicin has a melting point (
at the same temperature is due to dehydration. Doxorubicin has a melting point ( ) at about
) at about  as illustrated in literature [24], resulting in an endothermic peak in the DSC curve giving the evidence for crystal structure of the drug. The absence of native doxorubicin
as illustrated in literature [24], resulting in an endothermic peak in the DSC curve giving the evidence for crystal structure of the drug. The absence of native doxorubicin  peak on DSC curve of
peak on DSC curve of  nanoparticles shows that the drug in nanoparticles is in an amorphous phase. In other words, encapsulation process disrupts the native doxorubicin crystals, indicating that the encapsulation process is appreciable. However on the DSC curve of
nanoparticles shows that the drug in nanoparticles is in an amorphous phase. In other words, encapsulation process disrupts the native doxorubicin crystals, indicating that the encapsulation process is appreciable. However on the DSC curve of  , there is an exothermic peak centered at about
, there is an exothermic peak centered at about  . This may refer to the process of condensation of silanol (−Si–OH) groups that occurred at the surface of the silica particles and accompanied with the elimination of water molecule. This process supports the gathering of silica particles that cause the formation of new siloxane (−Si–O–Si−) bond at the interface. The presence of siloxane permits the more ordering of the product which demonstrate the appearance of the exothermic and peak negative enthalpy change [25]. This means that loading of DOX into silica nanoparticles lead to occurrence of dehydroxylation process at a lower temperature. The endothermic peak at about
. This may refer to the process of condensation of silanol (−Si–OH) groups that occurred at the surface of the silica particles and accompanied with the elimination of water molecule. This process supports the gathering of silica particles that cause the formation of new siloxane (−Si–O–Si−) bond at the interface. The presence of siloxane permits the more ordering of the product which demonstrate the appearance of the exothermic and peak negative enthalpy change [25]. This means that loading of DOX into silica nanoparticles lead to occurrence of dehydroxylation process at a lower temperature. The endothermic peak at about  may be due to removal of residual water molecules and other solvents present in pores.
may be due to removal of residual water molecules and other solvents present in pores.
3.4. Release of DOX using different mats
The in vitro release of DOX using the electrospun mats is studied by the immersion of the fibrous mat in PBS solution ( ) at
) at  . For comparison, the release of DOX by PLC-PEO/DOX and PLC-PEO/
. For comparison, the release of DOX by PLC-PEO/DOX and PLC-PEO/ in which
in which  is loaded by different methods;
is loaded by different methods;  and
and  are also investigated. As indicated in figure 6, all the electrospun mats show gradual increase of DOX release with time. The sample contains DOX loaded directly into the nanofiber. Curve in figure 6 indicates three stages of release profile when compared to other samples which exhibit two stages of release profile. The release profile using PLC-PEO/DOX indicates at first an initial rapid release within the first period of incubation, followed by a moderate increase in release with time and then almost constant release during the last periods of incubation. However the total amount of DOX release at the last period of incubation is 91.77%. This fast initial release could be attributed to the presence of DOX agglomerated on the surface of PCL-PEO nanofiber which leads to the direct exposure of DOX to the PBS that result in initial rapid release of DOX.
are also investigated. As indicated in figure 6, all the electrospun mats show gradual increase of DOX release with time. The sample contains DOX loaded directly into the nanofiber. Curve in figure 6 indicates three stages of release profile when compared to other samples which exhibit two stages of release profile. The release profile using PLC-PEO/DOX indicates at first an initial rapid release within the first period of incubation, followed by a moderate increase in release with time and then almost constant release during the last periods of incubation. However the total amount of DOX release at the last period of incubation is 91.77%. This fast initial release could be attributed to the presence of DOX agglomerated on the surface of PCL-PEO nanofiber which leads to the direct exposure of DOX to the PBS that result in initial rapid release of DOX.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. The cumulative DOX release profiles from nanofibrous mats prepared by different methods  , and
, and  (curve A, B, C, D and E, respectively).
(curve A, B, C, D and E, respectively).
Download figure:
Standard image High-resolution image{kind=link}
For the other samples in which  is loaded into the fiber by different methods, the release profile exhibits two stages of release profile; initial, slightly fast release, and secondly sustained release profile. The inhibition of the proliferation of the tumor cells through the early periods of implantation of fibrous mats loaded with anticancer drug is an important demand. This can be satisfied via the rapid initial drug release from the implanted mat. Also the sustained release of drug which follows initial rapid release provides an adequate concentration of anticancer drug over an extended period, this beneficial to avoid repeated administration of drug [3]. For the delivered samples, the DOX release rate is slower and the maximum amount of DOX release is less than that in case of using PLC-PEO-DOX [26].
is loaded into the fiber by different methods, the release profile exhibits two stages of release profile; initial, slightly fast release, and secondly sustained release profile. The inhibition of the proliferation of the tumor cells through the early periods of implantation of fibrous mats loaded with anticancer drug is an important demand. This can be satisfied via the rapid initial drug release from the implanted mat. Also the sustained release of drug which follows initial rapid release provides an adequate concentration of anticancer drug over an extended period, this beneficial to avoid repeated administration of drug [3]. For the delivered samples, the DOX release rate is slower and the maximum amount of DOX release is less than that in case of using PLC-PEO-DOX [26].
The release profile of  loaded into the nanofiber affected by the method of loading. As shown in curves B, C, D and E of figure 6, the release profile is higher (for both initial, and sustained stages) for sample in which
loaded into the nanofiber affected by the method of loading. As shown in curves B, C, D and E of figure 6, the release profile is higher (for both initial, and sustained stages) for sample in which  is loaded by M2 method as compared with M3, M4, M5 methods of loading. For example, the release of DOX at 33rd day (last day of incubation) is about 68.64% for this method and about 58.02%, 59.71%, 37.03% for the other three used methods respectively. M2 method results in the presence of
is loaded by M2 method as compared with M3, M4, M5 methods of loading. For example, the release of DOX at 33rd day (last day of incubation) is about 68.64% for this method and about 58.02%, 59.71%, 37.03% for the other three used methods respectively. M2 method results in the presence of  agglomerated on the surface of the nanofiber (as illustrated in FESEM micrographs) while the other methods of loading incorporate
agglomerated on the surface of the nanofiber (as illustrated in FESEM micrographs) while the other methods of loading incorporate  inside the nanofiber. This may be due to, DOX is released from
inside the nanofiber. This may be due to, DOX is released from  to the medium when present on the surface while when incorporated into the fiber, tends to be first released from the SiO2 matrix. Subsequently release is occurred from the polymer matrix to the medium [4]. While both M4 and M5 methods load
to the medium when present on the surface while when incorporated into the fiber, tends to be first released from the SiO2 matrix. Subsequently release is occurred from the polymer matrix to the medium [4]. While both M4 and M5 methods load  into the nanofiber, the release profile of DOX from core shell is slower. This may be due to the preparation by core shell which results in the formation of core which contains dispersed
into the nanofiber, the release profile of DOX from core shell is slower. This may be due to the preparation by core shell which results in the formation of core which contains dispersed  and also thick sheath formed of polymer which means that DOX undergoes more difficulties in steps when released to the medium [27].
and also thick sheath formed of polymer which means that DOX undergoes more difficulties in steps when released to the medium [27].
4. Conclusion
PCL-PEO fiber mats are prepared via electrospinning method as a carrier for DOX and sol gel derived  nanoparticles. Morphological observations (FESEM micrographs) reveal that the incorporation method is greatly affects the distribution of
nanoparticles. Morphological observations (FESEM micrographs) reveal that the incorporation method is greatly affects the distribution of  nanoparticles within the fibrous structure of the polymeric mats. Moreover, in-vitro drug release results in reflectance of the capability of minimal side effects of DOX using encapsulation of drug loading nanoparticles by electrospun fibers. The results reported here provide useful information for the fabrication of DOX releasing implants for clinical treatment of cancer because of high potential of control and localization of a drug dose.
nanoparticles within the fibrous structure of the polymeric mats. Moreover, in-vitro drug release results in reflectance of the capability of minimal side effects of DOX using encapsulation of drug loading nanoparticles by electrospun fibers. The results reported here provide useful information for the fabrication of DOX releasing implants for clinical treatment of cancer because of high potential of control and localization of a drug dose.
Acknowledgments
The authors gratefully acknowledge the support from the faculty of science, Al-Azhar University and also the reinforce from the National Research Center of Egypt (NRC).